CIGAR
Eine CIGAR (Concise Idiosyncratic Gapped Alignment Report) String besteht aus einer Reihe von Operationslängen sowie den Operationen, die beschreiben, wie genau ein Read an die Referenzsequenz angeglichen wurde (Abbildung 1).
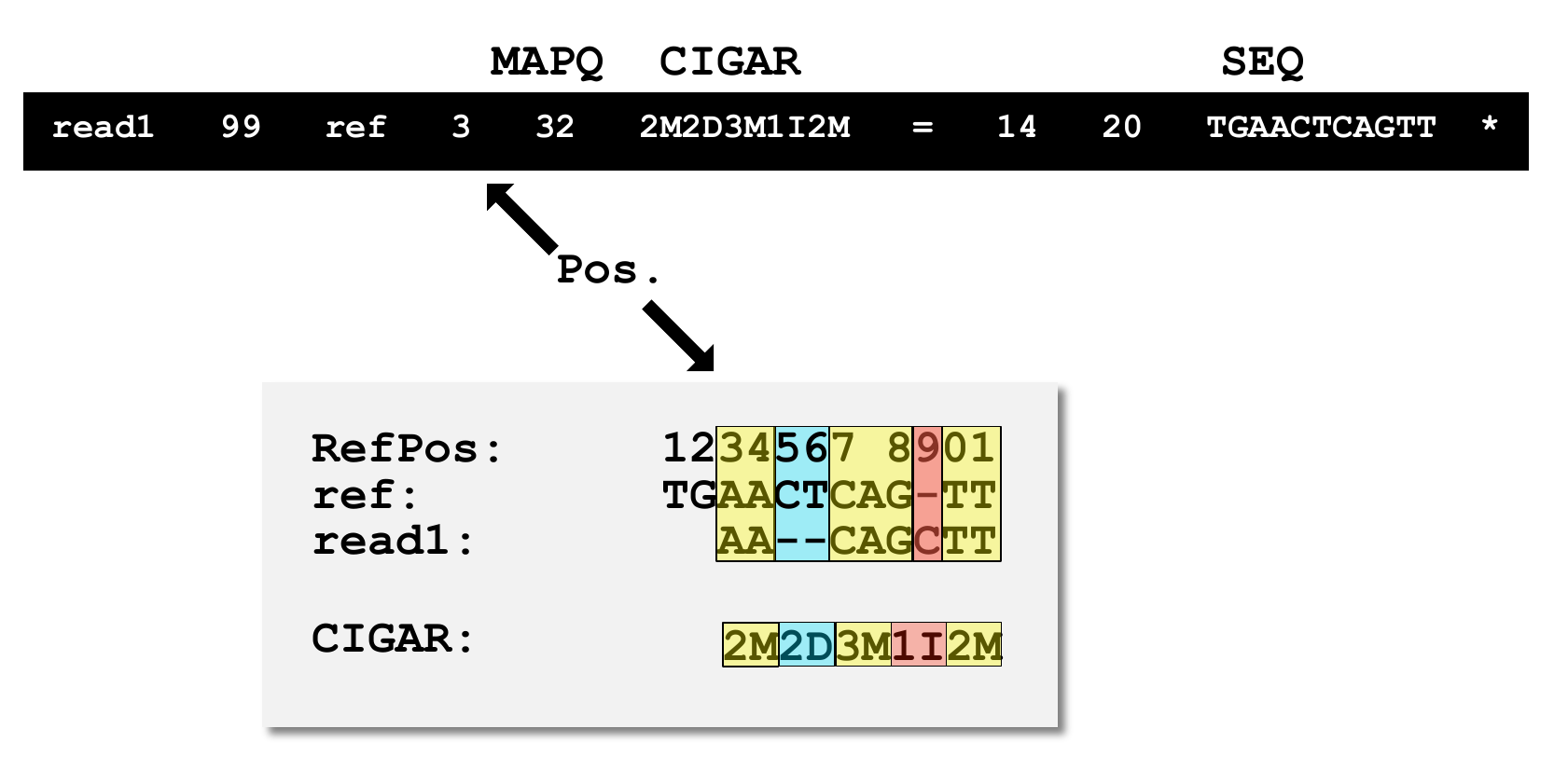
Die drei wichtigsten CIGAR-Operationen sind M (Match/Mismatch), I (Insertion) und D (Deletion), aber aber es gibt noch sechs weitere Operationen, von denen einige im Folgenden erläutert werden. Alle CIGAR Operationen sind in Tabelle 1 zusammengefasst. Eine CIGAR-Zeichenkette kann man sich als eine Reihe von Operationslängen plus die CIGAR-Operation, die eine abstrakte und kompakte Darstellung beliebiger Ausrichtungen ermöglicht. Betrachten Sie beispielsweise das folgende Alignment, in dem die Abfragesequenz (Q) an die Referenzsequenz (R) angeglichen wird, beginnend an Position 7 der Referenzsequenz.
AGCATGTTAGATAA--GATAGCTGG R
|| ||||| |||| ||||
------TTGGATAAAGGATA-CTGG Q
Es gibt 8 anfängliche Übereinstimmungen, dann eine Einfügung
von 2 Nukleotiden in die Abfrage, dann vier weitere Übereinstimmungen,
dann eine Löschung von einer Base in der Abfrage und dann vier weitere
Übereinstimmungen. Wir können dies als 8M2I4M1D4M darstellen.
Man beachte, dass die dritte Base der Abfragesequenz (G) sich tatsächlich von der Base der Referenzsequenz (A) unterscheidet, an die sie angeglichen wurde. Nichtsdestotrotz handelt es sich um eine "Alignment match" (Ausrichtungsübereinstimmung) (M), obwohl es keine Sequenzübereinstimmung ist.
| Operation (Op) | Beschreibung |
|---|---|
| M | Match (Sequenzübereinstimmung oder Fehlübereinstimmung, keiner Insertion oder Deletion) |
| I | Insertion (zusätzliche Nicht-Referenzbase) |
| D | Deletion (Referenzbase fehlt in der Lesung) |
| N | skipped region=übersprungener Bereich aus der Referenz |
| S | soft clipping (geclippte Sequenzen noch in SEQ vorhanden) |
| H | hartes Clipping (geclippte Sequenzen nicht in SEQ vorhanden) |
| P | padding (stille Löschung aus gepolsterter Referenz) |
| = | sequence match: Sequenzübereinstimmung |
| X | sequence mismatch=Sequenz-Fehlanpassung |
Übung 1
Nehmen Sie eine Referenz- und eine Querysequenz sowie den CIGAR-String, der das Alignment spezifiziert (z.B. von Abb. 1). In unserem Beispiel wären die drei Argumente also
- Query (read_4): GCATCTGTTG
- Ref: GCATACTGTTG
- CIGAR: 4M1D6M
Das Ziel ist es, eine Funktion zu schreiben, welche das entsprechende Alignment ausgibt, also in unserem Beispiel
GCAT-CTGTTG |||| |||||| GCATACTGTTG
Übung 2
Probieren Sie Ihre Funktion mit "realistischen" BAM-Dateien -- wir haben eine TODO -- "kleine" BAM-Datei erstellen.