SAM-Grundlagen
Wir werden das grundlegende SAM-Format anhand eines Beispiels erläutern, bei dem fünf Reads der Länge 10 auf eine Referenzsequenz der Länge 40 abgebildet werden (Abbildung 1).
Lesen Sie die Ausführungen zum SAM-Format und bearbeiten Sie die Übung am Ende der Seite.
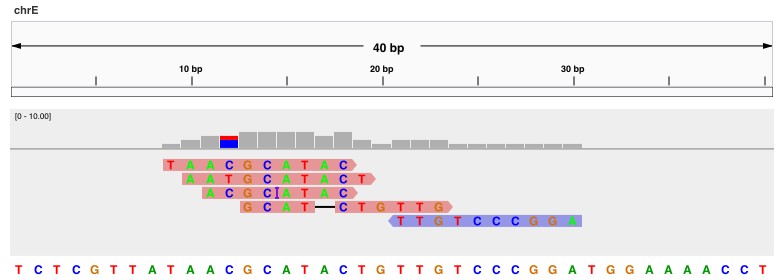

SAM-Dateien bestehen aus einem optionalen Header-Abschnitt, gefolgt von einem Alignment-Abschnitt. Die Kopfzeilen beginnen mit `@'. Im Alignment-Abschnitt der SAM-Datei stellt jede Zeile ein Alignment für einen Read dar und umfasst mindestens 11 Pflichtfelder (Tabelle 1).
| Col | Feld | Beschreibung | Beispiel |
|---|---|---|---|
| 1 | QNAME |
Query template NAME | read_1 |
| 2 | FLAG |
Bitweise Flag | 0 |
| 3 | RNAME |
Name der Referenzsequenz | chrE |
| 4 | POS |
Linke Kartierungsposition (1-basiert) | 11 |
| 5 | MAPQ |
MAPping Qualität | 37 |
| 6 | CIGAR |
CIGAR String | 10M |
| 7 | RNEXT |
Referenzname des Mate-Reads oder NEXT read | * |
| 8 | PNEXT |
Position des Mate-Reads oder NEXT read | 0 |
| 9 | TLEN |
Beobachtete Template-Länge (LENgth) | 0 |
| 10 | SEQ |
Segment-SEQuenz | ACGCATACTG |
| 11 | QUAL |
Basen-QUALitätsstring | DIGAFHHBCA |
Jede Zeile im Alignmentsabschnitt einer SAM-Datei umfasst 11 Pflichtfelder.
Das SAM/BAM-Format soll so allgemein wie möglich sein und verwendet das Wort
template für DNA-Fragmente (englisch: fragments, inserts).
Das SAM-Format verwendet das Wort
segment, um sich auf eine zusammenhängende Sequenz oder
Teilsequenz zu beziehen; zum Beispiel können die beiden Reads eines
Lesepaares als zwei Segmente bezeichnet werden.
Das Feld QNAME speichert den Namen der Abfrage-(query)-Sequenz,
in der Regel ein Read.1
POS gibt die Position auf dem Chromosom an, an der der Read
ausgerichtet wurde. Das Feld FLAG wird weiter unten erklärt.
Das Feld MAPQ stellt die
Mappingqualität, die vom Aligner zugewiesen wurde, und spiegelt das Vertrauen
wider, mit dem der Read auf die angegebene Position abgebildet werden konnte.
Die CIGAR-Zeichenkette bietet eine kompakte
kompakte Darstellung des Alignments, die im Folgenden erläutert wird.
Die Felder RNEXT,
PNEXT und TLEN werden nur für Paired-End-Reads verwendet.
Daher wird bei Single-End
Reads RNEXT auf ``*'' (für not-applicable) gesetzt.
PNEXT und TLEN werden auf 0 gesetzt. Schließlich enthält
das Feld SEQ die Nukleotidsequenz des Reads und das Feld QUAL
zeigt die Basenqualitätswerte für jede Position an.
Paired-End-Sequenzen
In Abbildung 2 werden zwei Paare
bestehend aus vier Reads der Länge 10 werden auf eine Referenzsequenz der Länge 40 abgebildet. Beachten Sie, dass
Mitglieder desselben Paars haben denselben QNAME und werden auf verschiedene Stränge abgebildet. Read 1 wird dem
dem Vorwärtsstrang an Position 2 zugeordnet und bildet ein Paar mit dem Read, der dem Rückwärtsstrang an Position
Strang an Position 27 zugeordnet ist. RNEXT wird auf = gesetzt, da der zweite Read des Paares
des Paares auf dieselbe Referenz, d. h. chrE, abgebildet wird, und PNEXT wird auf 27 gesetzt, weil der
Reverse-Read auf diese Position abgebildet wird (der Reverse-Read ist an den Positionen 27-36 ausgerichtet).
Daher wird TLEN auf 35 gesetzt, denn dies ist der Abstand zwischen der äußersten linken und der äußersten rechten
der am weitesten rechts gelegenen Base der beiden Reads. Der Eintrag TLEN für den Reverse-Read wird mit
einem Minuszeichen dargestellt.
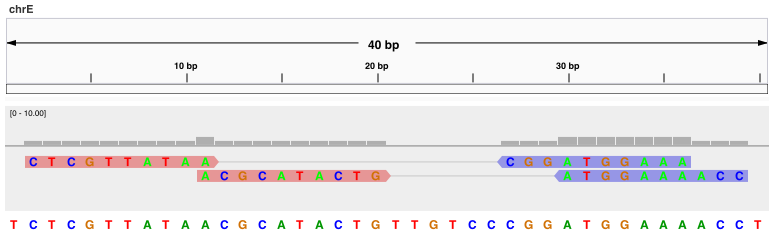
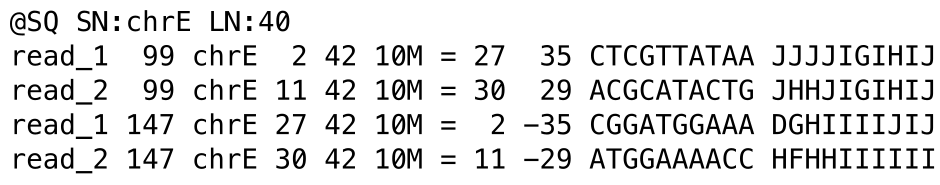
Für Paired-End-Reads werden andere bitweise Flags verwendet als für Single-End-Reads. Die Flags 99 und 147 zeigen an, dass beide Mitglieder des Paares korrekt auf den Vorwärts- und den Rückwärtsstrang gemappt wurden.
-
Segmente mit demselben
QNAMEstammen von der gleichen Vorlage, d. h. Reads eines Read-Paares haben normalerweise den gleichenQNAME. ↩