Clipped Reads
Wichtige Mapping-Tools wie BWA versuchen, einen Teil eines Reads zu kartieren, wenn sie den Read nicht in voller Länge auf das Referenzgenom abbilden können. In diesen Fällen kann der nicht zuzuordnende Teil durch einen Prozess namens
Mit soft-clipping, das in der CIGAR-Zeichenkette mit einem >S angezeigt wird, werden die abgeschnittenen Sequenzbasen nicht aus der SEQ-Zeichenkette entfernt, aber nicht von Varianten-Callern verwendet und nicht in Viewern wie IGV (Integrativer Genomik-Viewer)angezeigt.
Das Hard-Clipping (H) ähnelt dem Soft-Clipping (S), unterscheidet sich aber dadurch, dass die hart geclippte Teilsequenz nicht im Alignment-Datensatz enthalten ist (Abbildung 1).1
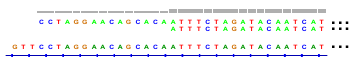
Ref: GTTCCTAGGAACAGCACAATTTCTAGATACAATCAT Read1: CCTAGGAACAGCACAATTTCTAGATACAATCAT Read2: ggtcacatgattgtATTTCTAGATACAATCAT
Das entsprechende Alignment von Read1 und Read2 mit Ref. Die soft-clipped (nicht ausrichtbaren) Basen von Read2 sind in Kleinbuchstaben dargestellt.
-
BWA verwendet das Soft-Clipping für das primäre Alignment, damit die ursprünglichen Rohdaten bei Bedarf aus der BAM-Datei regeneriert werden können. Für die sekundären Alignments ist dies nicht notwendig, daher verwendet BWA Hard Clipping, um Speicherplatz zu sparen. ↩